Bulk Enrichment Analysis#
Bulk RNA-seq yields many molecular readouts that are hard to interpret by themselves. One common approach to summarize this data is to compute enrichment scores using predefined gene sets based on prior biological knowledge.
This notebook demonstrates the use of decoupler to infer transcription factor (TF)
and pathway enrichment scores from a bulk RNA-seq dataset of a human cell line [YLG+20].
The dataset includes six samples of hepatic stellate cells (HSCs), three of which were activated with the cytokine Transforming Growth Factor beta (TGF-β). It is publicly available at GEO (GSE151251).
Loading Packages#
import numpy as np
import scanpy as sc
import decoupler as dc
sc.set_figure_params(figsize=(3, 3), frameon=False)
Loading The Dataset#
adata = dc.ds.hsctgfb()
adata
AnnData object with n_obs × n_vars = 6 × 58674
obs: 'condition', 'sample_id'
layers: None
The obtained anndata.AnnData consist of raw integer transcript counts for six different samples
(three controls, three treatments) with measurements for ~60k genes.
We can inspect the sample metadata stored in anndata.AnnData.obs.
adata.obs
| condition | sample_id | |
|---|---|---|
| 25_HSCs-Ctrl1 | control | 25 |
| 26_HSCs-Ctrl2 | control | 26 |
| 27_HSCs-Ctrl3 | control | 27 |
| 31_HSCs-TGFb1 | treatment | 31 |
| 32_HSCs-TGFb2 | treatment | 32 |
| 33_HSCs-TGFb3 | treatment | 33 |
And visualized.
dc.pl.obsbar(adata=adata, y="condition", hue="condition", figsize=(3, 3))
Although this plot is simple in the current example, it can be useful in more complex experimental designs.
Preprocessing#
Quality Control#
Before proceeding, we need to ensure that our data meets basic quality control standards. In transcriptomics, some genes may be poorly profiled and should be excluded from analysis.
To filter genes, we follow the approach used in the filterByExpr function from
edgeR [RMS09].
This method retains genes that meet two criteria:
A minimum total read count across all samples (
min_total_count)A minimum read count in a minimum number of samples (
min_count)
We can visualize how many genes are retained under different filtering thresholds.
Feel free to adjust the parameters of decoupler.pl.filter_by_expr to explore
how they affect gene retention.
dc.pl.filter_by_expr(
adata=adata,
group="condition",
min_count=10,
min_total_count=15,
large_n=10,
min_prop=0.7,
)
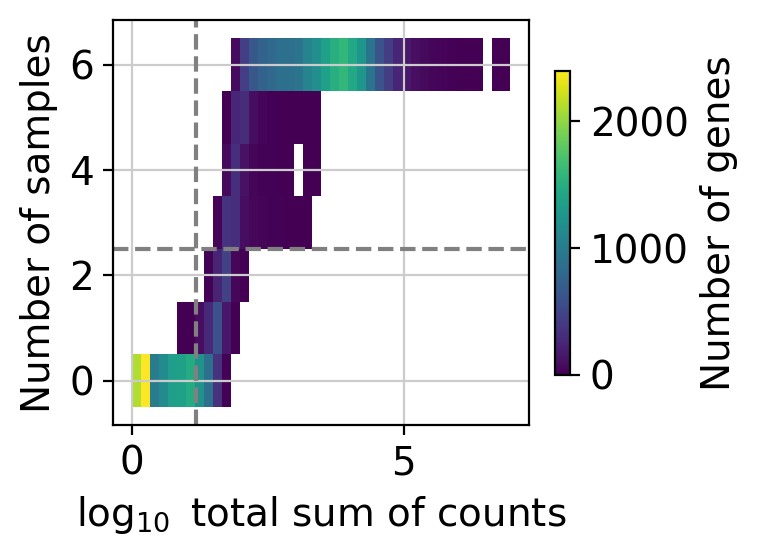
Here, we visualize the distribution of genes by their total count across all samples and the number of samples in which they are expressed. The dashed lines represent the current filtering thresholds, only genes in the upper-right corner will be retained.
While filtering thresholds are somewhat arbitrary, a common strategy is to look for bimodal distributions and set thresholds that separate low-expression (potentially noisy) genes from those with more robust expression.
In this example, the default settings strike a balance by retaining a substantial number of genes while filtering out likely noise.
Note
Changing the value of min_count will significantly affect the distribution of the “Number of samples”
but will not directly change its threshold. To adjust this threshold, either to lower or raise it,
you’ll need to modify the group, large_n, and min_prop parameters.
Note
Filtering thresholds can vary greatly between datasets, so manual assessment is important.
Once you’re satisfied with the threshold parameters, you can proceed with the actual filtering
with the function decoupler.pp.filter_by_expr.
dc.pp.filter_by_expr(
adata=adata,
group="condition",
min_count=10,
min_total_count=15,
large_n=10,
min_prop=0.7,
)
Variability Exploration#
Before performing any test between groups, it is a good practice to explore the variability of the omics profiles.
This involves some basic preprocessing followed by principal component analysis (PCA).
# Store raw counts in layers
adata.layers["counts"] = adata.X.copy()
# Normalize, scale and compute pca
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
sc.pp.scale(adata, max_value=10)
sc.tl.pca(adata)
# Return raw counts to X
dc.pp.swap_layer(adata=adata, key="counts", inplace=True)
After computing the PCs, associations or correlations between each inferred PC and the variables in the metadata can be tested, depending on whether the variables are categorical or continuous.
This type of analysis is applicable to any dimensionality reduction method, such as factors derived from non-negative matrix factorization.
dc.tl.rankby_obsm(adata, key="X_pca")
The importance of each principal component (based on its explained variance ratio) and its associations with metadata variables can then be visualized.
sc.pl.pca_variance_ratio(adata)
dc.pl.obsm(adata=adata, return_fig=True, nvar=5, titles=["PC scores", "Adjusted p-values"], figsize=(6, 3))
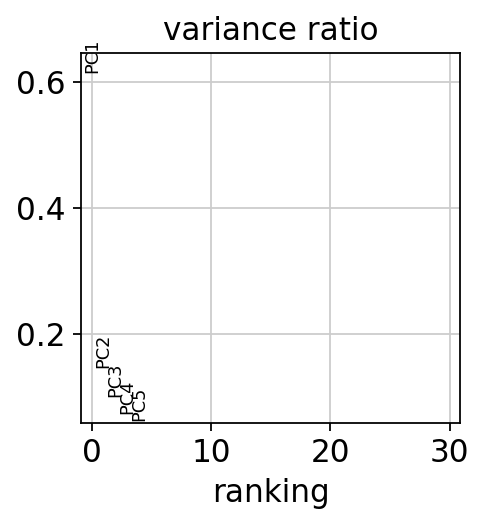
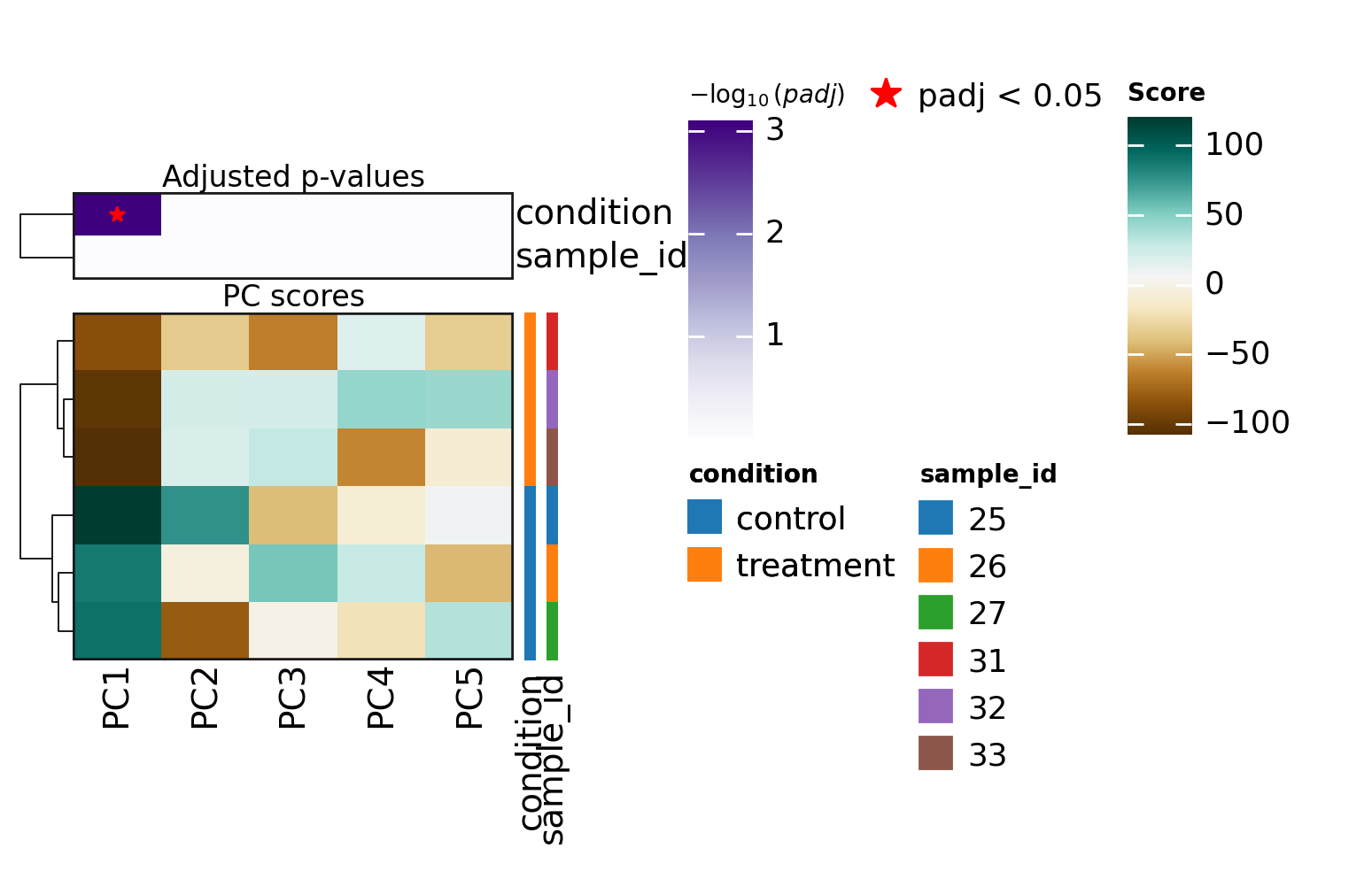
In this dataset, PC1 appears to explain the largest proportion of variance and is associated with the metadata variable “condition”, which is a good indication that the experiment worked.
Metadata variables associated with PCs that capture a substantial amount of variance are important and should be accounted for as relevant covariates in downstream differential expression analysis when possible.
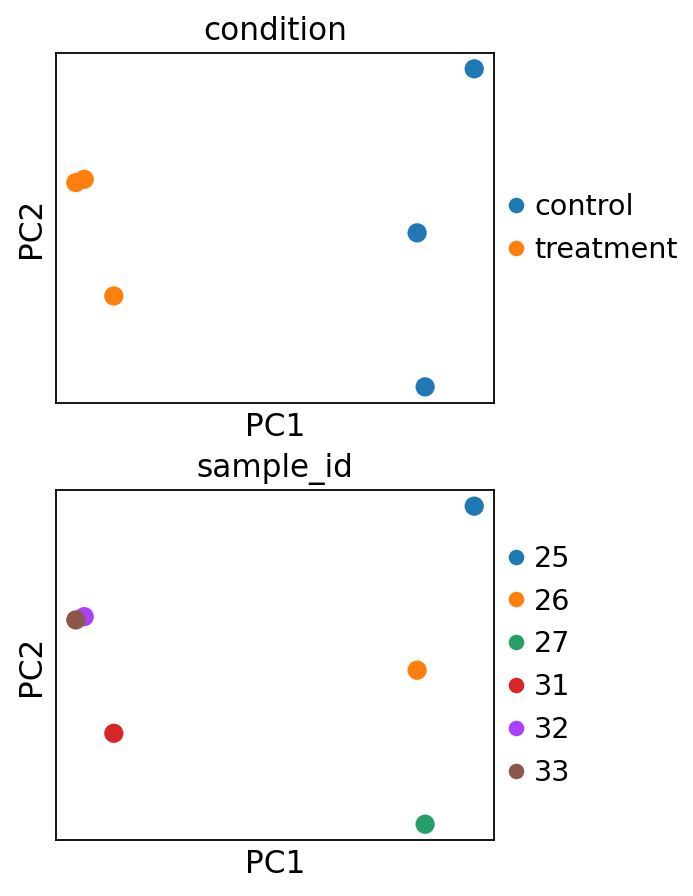
The principal components can also be directly visualized, colored by these metadata variables.
sc.pl.pca(
adata,
color=["condition", "sample_id"],
ncols=1,
size=300,
frameon=True,
)
Differential Expression Analysis#
To identify the genes that change most between treatment and control, we perform differential expression analysis (DEA). In this example, we use a simple experimental design that compares gene expression in treated cells versus controls.
We will use the Python implementation of the DESeq2 framework [MTCA23], though other tools like limma
[RPW+15] or edgeR [RMS09] could also be used.
For a deeper understanding of how pyDESeq2 works, refer to its
official documentation.
Note that more complex experimental designs can be accommodated by adding additional factors to the design formula.
# Import DESeq2
from pydeseq2.dds import DeseqDataSet, DefaultInference
from pydeseq2.ds import DeseqStats
# Build DESeq2 object
inference = DefaultInference(n_cpus=8)
dds = DeseqDataSet(
adata=adata,
design="~condition",
refit_cooks=True,
inference=inference,
)
# Compute LFCs
dds.deseq2()
# Extract contrast between conditions
stat_res = DeseqStats(dds, contrast=["condition", "treatment", "control"], inference=inference)
# Compute Wald test
stat_res.summary()
Using None as control genes, passed at DeseqDataSet initialization
Log2 fold change & Wald test p-value: condition treatment vs control
baseMean log2FoldChange lfcSE stat pvalue \
WASH7P 10.350605 -0.010706 0.650918 -0.016447 0.986878
MIR6859-1 10.115837 0.001174 0.656587 0.001788 0.998573
RP11-34P13.7 45.735113 0.078681 0.324022 0.242827 0.808139
RP11-34P13.8 29.500549 -0.064734 0.393110 -0.164670 0.869203
CICP27 106.044923 0.151057 0.222763 0.678106 0.497705
... ... ... ... ... ...
MT-ND6 17915.987884 -0.434814 0.278711 -1.560087 0.118739
MT-TE 1281.386013 -0.332033 0.288055 -1.152672 0.249045
MT-CYB 54959.449877 -0.312805 0.286884 -1.090354 0.275557
MT-TT 204.708230 -0.485499 0.220416 -2.202654 0.027619
MT-TP 345.083555 -0.460288 0.161650 -2.847430 0.004407
padj
WASH7P 0.991401
MIR6859-1 0.999344
RP11-34P13.7 0.875253
RP11-34P13.8 0.916955
CICP27 0.634945
... ...
MT-ND6 0.210485
MT-TE 0.379777
MT-CYB 0.410724
MT-TT 0.061263
MT-TP 0.012197
[19510 rows x 6 columns]
# Extract results
results_df = stat_res.results_df
results_df
| baseMean | log2FoldChange | lfcSE | stat | pvalue | padj | |
|---|---|---|---|---|---|---|
| WASH7P | 10.350605 | -0.010706 | 0.650918 | -0.016447 | 0.986878 | 0.991401 |
| MIR6859-1 | 10.115837 | 0.001174 | 0.656587 | 0.001788 | 0.998573 | 0.999344 |
| RP11-34P13.7 | 45.735113 | 0.078681 | 0.324022 | 0.242827 | 0.808139 | 0.875253 |
| RP11-34P13.8 | 29.500549 | -0.064734 | 0.393110 | -0.164670 | 0.869203 | 0.916955 |
| CICP27 | 106.044923 | 0.151057 | 0.222763 | 0.678106 | 0.497705 | 0.634945 |
| ... | ... | ... | ... | ... | ... | ... |
| MT-ND6 | 17915.987884 | -0.434814 | 0.278711 | -1.560087 | 0.118739 | 0.210485 |
| MT-TE | 1281.386013 | -0.332033 | 0.288055 | -1.152672 | 0.249045 | 0.379777 |
| MT-CYB | 54959.449877 | -0.312805 | 0.286884 | -1.090354 | 0.275557 | 0.410724 |
| MT-TT | 204.708230 | -0.485499 | 0.220416 | -2.202654 | 0.027619 | 0.061263 |
| MT-TP | 345.083555 | -0.460288 | 0.161650 | -2.847430 | 0.004407 | 0.012197 |
19510 rows × 6 columns
The results can be visualized using a volcano plot.
dc.pl.volcano(
data=results_df,
x="log2FoldChange",
y="padj",
top=10,
)
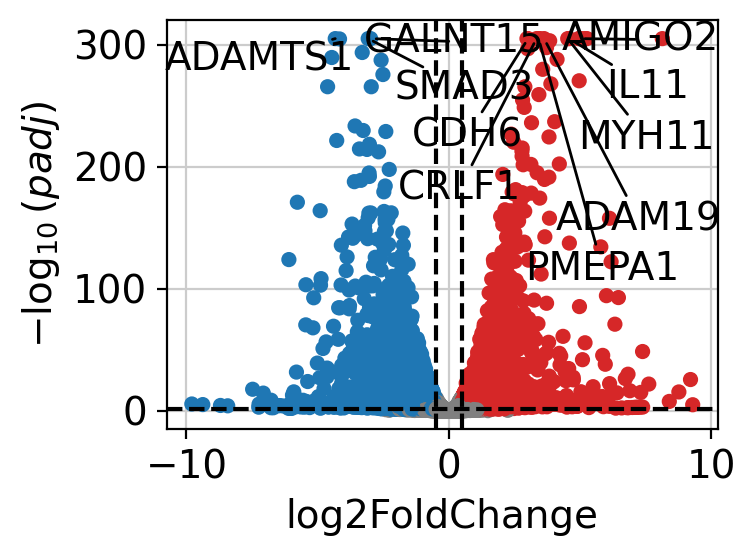
After performing DEA, we can use the resulting gene-level statistics for enrichment analysis.
While any statistic can be used, we recommend using t-values rather than log2FCs,
as t-values account for the significance of the change.
We will transform the obtained t-values, stored in the column stat, into a wide matrix
format so that it can be used with decoupler.
data = results_df[["stat"]].T.rename(index={"stat": "treatment.vs.control"})
data
| WASH7P | MIR6859-1 | RP11-34P13.7 | RP11-34P13.8 | CICP27 | FO538757.2 | AP006222.2 | RP4-669L17.10 | MTND1P23 | MTND2P28 | ... | MT-ND4 | MT-TH | MT-TS2 | MT-TL2 | MT-ND5 | MT-ND6 | MT-TE | MT-CYB | MT-TT | MT-TP | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| treatment.vs.control | -0.016447 | 0.001788 | 0.242827 | -0.16467 | 0.678106 | -1.646572 | 2.045171 | -0.376314 | -1.992066 | -0.49685 | ... | -1.434342 | 0.757892 | 1.141412 | 1.169607 | -1.241004 | -1.560087 | -1.152672 | -1.090354 | -2.202654 | -2.84743 |
1 rows × 19510 columns
Enrichment analysis#
Enrichment analysis tests whether a specific set of omics features is “overrepresented” or “coordinated” in the measured data compared to a background distribution. These sets are predefined based on existing biological knowledge and may vary depending on the omics technology used.
Enrichment analysis requires the use of an enrichment method, and several options are
available.
In the original manuscript of decoupler [BiMVSB+22], we benchmarked multiple
methods and found that the univariate linear model (ulm) outperformed the others.
Therefore, we will use ulm in this vignette.
The scores from decoupler.mt.ulm should be interpreted such that larger magnitudes
indicate greater significance, while the sign reflects whether the features in the set are
overrepresented (positive) or underrepresented (negative) compared to the background.
Transcription factor scoring from gene regulatory networks#
Transcription factors (TFs) are genes that, once translated into proteins, bind to DNA and regulate the expression of other genes by either promoting or inhibiting their transcription. Gene Regulatory Networks (GRNs) capture these TF-gene interactions and can be constructed from prior knowledge or inferred from omics data. The fundamental unit of a GRN is a TF and its associated target genes, collectively known as a regulon. Each regulon functions as a gene set in enrichment analysis.
Although TFs are measured in transcriptomic data, their transcript levels often do not reflect their actual activity in a given cell. Instead, scoring TFs through enrichment analysis based on the expression of their target genes provides a more accurate representation of their regulatory activity [BiMWMullerD+23].
CollecTRI network#
CollecTRI is a comprehensive resource containing a curated collection of TFs and their transcriptional targets compiled from 12 different resources [MullerDTV+23]. This collection provides an increased coverage of transcription factors and a superior performance in identifying perturbed TFs compared to other literature based GRNs such as DoRothEA [GAHI+19]. Similar to DoRothEA, interactions are weighted by their mode of regulation (activation or inhibition).
In this tutorial we will use the human version but other organisms are available.
We can use decoupler to retrieve it from the OmniPath server.
Note
In this tutorial we use the network CollecTRI, but we could use any other GRN coming from an inference method such as CellOracle, pySCENIC or SCENIC+.
collectri = dc.op.collectri(organism="human")
collectri
| source | target | weight | resources | references | sign_decision | |
|---|---|---|---|---|---|---|
| 0 | MYC | TERT | 1.0 | DoRothEA-A;ExTRI;HTRI;NTNU.Curated;Pavlidis202... | 10022128;10491298;10606235;10637317;10723141;1... | PMID |
| 1 | SPI1 | BGLAP | 1.0 | ExTRI | 10022617 | default activation |
| 2 | SMAD3 | JUN | 1.0 | ExTRI;NTNU.Curated;TFactS;TRRUST | 10022869;12374795 | PMID |
| 3 | SMAD4 | JUN | 1.0 | ExTRI;NTNU.Curated;TFactS;TRRUST | 10022869;12374795 | PMID |
| 4 | STAT5A | IL2 | 1.0 | ExTRI | 10022878;11435608;17182565;17911616;22854263;2... | default activation |
| ... | ... | ... | ... | ... | ... | ... |
| 42985 | NFKB | hsa-miR-143-3p | 1.0 | ExTRI | 19472311 | default activation |
| 42986 | AP1 | hsa-miR-206 | 1.0 | ExTRI;GEREDB;NTNU.Curated | 19721712 | PMID |
| 42987 | NFKB | hsa-miR-21 | 1.0 | ExTRI | 20813833;22387281 | default activation |
| 42988 | NFKB | hsa-miR-224-5p | 1.0 | ExTRI | 23474441;23988648 | default activation |
| 42989 | AP1 | hsa-miR-144-3p | 1.0 | ExTRI | 23546882 | default activation |
42990 rows × 6 columns
Scoring#
We can easily compute TF scores by running the ulm method.
# Run
tf_acts, tf_padj = dc.mt.ulm(data=data, net=collectri)
# Filter by sign padj
msk = (tf_padj.T < 0.05).iloc[:, 0]
tf_acts = tf_acts.loc[:, msk]
tf_acts
| APEX1 | ARID1A | ARID4B | ARNT | ASXL1 | ATF6 | BARX2 | BCL11A | BCL11B | BMAL2 | ... | TBX10 | TCF21 | TFAP2C | TP53 | VEZF1 | WWTR1 | ZBTB33 | ZEB2 | ZNF804A | ZXDC | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| treatment.vs.control | -2.659163 | -3.917493 | -2.807218 | 3.171876 | 3.745517 | 2.706447 | 5.3575 | 2.648758 | -4.203275 | 3.868107 | ... | -3.703849 | 6.161999 | -2.579122 | -3.523291 | 4.207675 | -3.968219 | 3.236389 | 5.789579 | 3.364015 | -2.733644 |
1 rows × 143 columns
The obtained scores for the most active and inactive TFs can be visualized as follows.
dc.pl.barplot(data=tf_acts, name="treatment.vs.control", top=25, figsize=(3, 5))
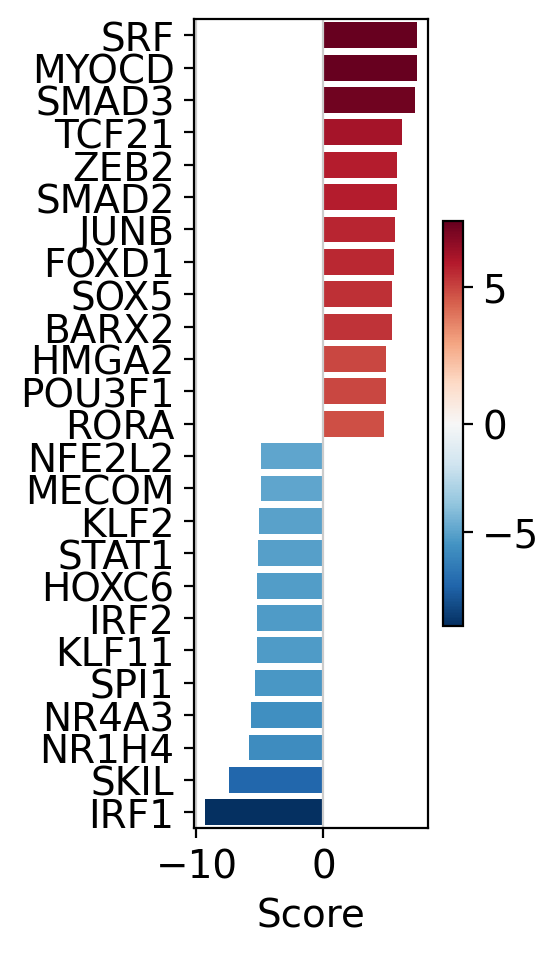
SRF, MYOCD and SMAD3 appear to be the most activated TFs in this treatment, while IRF1, SKIL and NR1H4 appear to be inactivated.
A network of selected TFs (top and bottom ranked by activity) can also be visualized, with nodes colored by TF activity and target gene-level statistics.
dc.pl.network(
net=collectri,
data=data,
score=tf_acts,
sources=["SRF", "MYOCD", "IRF1", "SKIL"],
targets=10,
figsize=(5, 5),
vcenter=True,
by_abs=True,
size_node=10,
t_label="Gene-level\nstatistic",
)
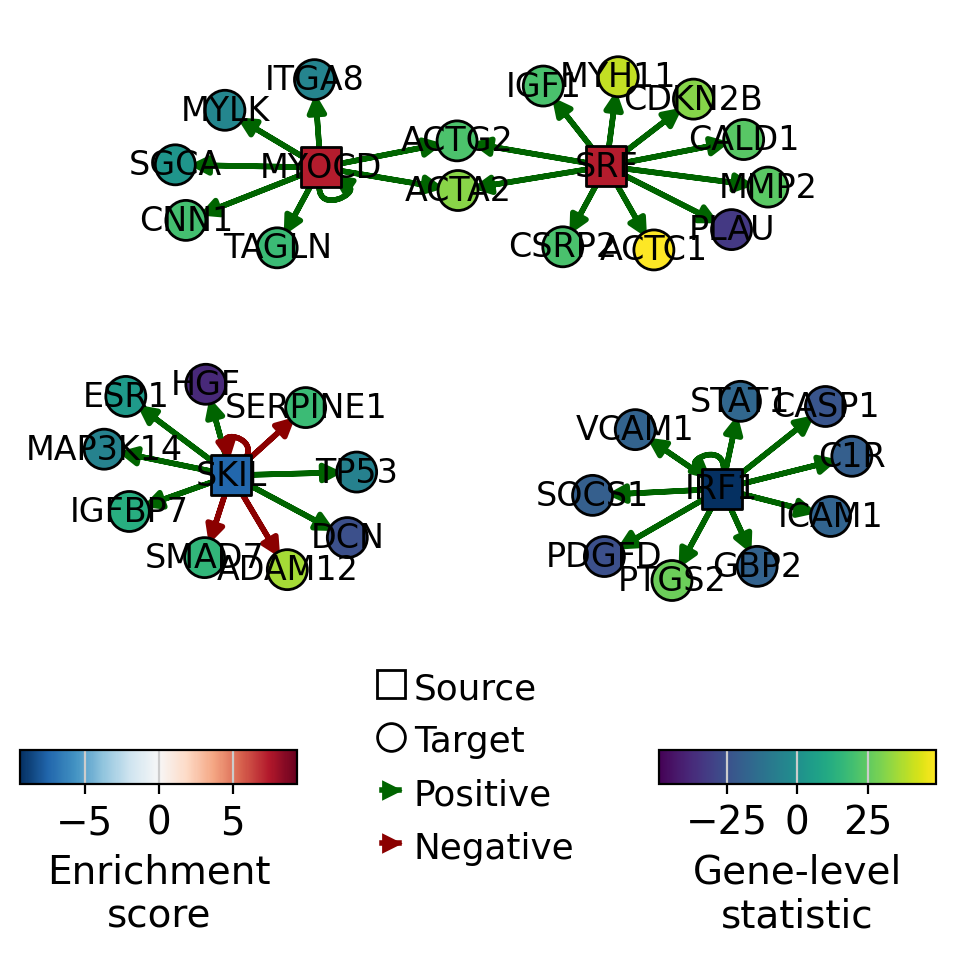
SRF appears to be active in treated cells, as its positively regulated targets are upregulated.
If needed, we can also look at a volcano plot of the target genes.
dc.pl.volcano(
data=results_df,
x="log2FoldChange",
y="pvalue",
net=collectri,
name="SRF",
top=10,
)
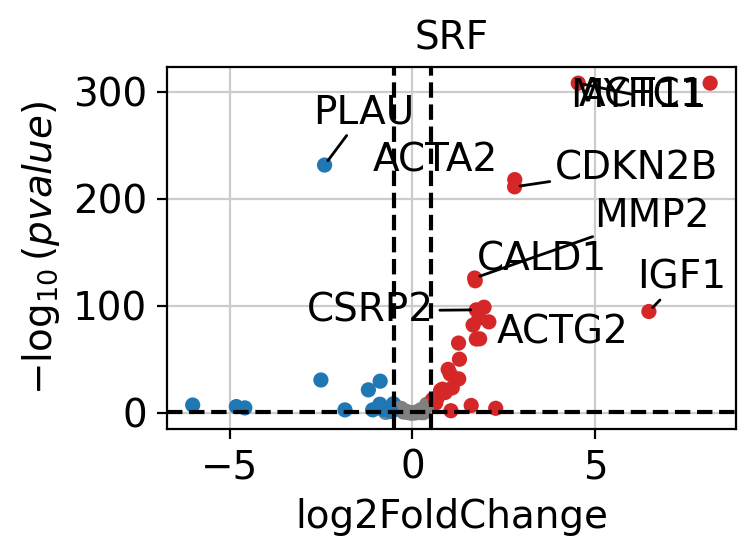
Pathway Scoring#
The same approach used for TF scoring can also be applied to pathways. Numerous
databases provide curated pathway gene sets, with one of the most well-known being MSigDB, which
includes several collections [LSP+11].
These and many other resources can be accessed using the function decoupler.op.resource().
To view the list of available databases, use decoupler.op.show_resources().
PROGENy Pathway Genes#
PROGENy is a comprehensive resource that provides a curated collection of pathways and their target genes, along with weights for each interaction [SKK+18].
Below is a brief description of each pathway:
Androgen: involved in the growth and development of the male reproductive organs
EGFR: regulates growth, survival, migration, apoptosis, proliferation, and differentiation in mammalian cells
Estrogen: promotes the growth and development of the female reproductive organs
Hypoxia: promotes angiogenesis and metabolic reprogramming when O2 levels are low
JAK-STAT: involved in immunity, cell division, cell death, and tumor formation
MAPK: integrates external signals and promotes cell growth and proliferation
NFkB: regulates immune response, cytokine production and cell survival
p53: regulates cell cycle, apoptosis, DNA repair and tumor suppression
PI3K: promotes growth and proliferation
TGFb: involved in development, homeostasis, and repair of most tissues
TNFa: mediates haematopoiesis, immune surveillance, tumour regression and protection from infection
Trail: induces apoptosis
VEGF: mediates angiogenesis, vascular permeability, and cell migration
WNT: regulates organ morphogenesis during development and tissue repair
This is how to access to it.
progeny = dc.op.progeny(organism="human")
progeny
| source | target | weight | padj | |
|---|---|---|---|---|
| 0 | Androgen | TMPRSS2 | 11.490631 | 2.384806e-47 |
| 1 | Androgen | NKX3-1 | 10.622551 | 2.205102e-44 |
| 2 | Androgen | MBOAT2 | 10.472733 | 4.632376e-44 |
| 3 | Androgen | KLK2 | 10.176186 | 1.944410e-40 |
| 4 | Androgen | SARG | 11.386852 | 2.790210e-40 |
| ... | ... | ... | ... | ... |
| 62416 | p53 | ENPP2 | 2.771405 | 4.993215e-02 |
| 62417 | p53 | ARRDC4 | 3.494328 | 4.996747e-02 |
| 62418 | p53 | MYO1B | -1.148057 | 4.997905e-02 |
| 62419 | p53 | CTSC | -1.784693 | 4.998864e-02 |
| 62420 | p53 | NAA50 | -1.435013 | 4.998884e-02 |
62421 rows × 4 columns
Scoring#
We can easily compute pathway scores by running the ulm method.
# Run
pw_acts, pw_padj = dc.mt.ulm(data=data, net=progeny)
# Filter by sign padj
msk = (pw_padj.T < 0.05).iloc[:, 0]
pw_acts = pw_acts.loc[:, msk]
pw_acts
| Androgen | EGFR | Estrogen | Hypoxia | JAK-STAT | MAPK | NFkB | PI3K | TGFb | TNFa | VEGF | p53 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| treatment.vs.control | 4.709702 | 17.26656 | 10.928969 | 5.728908 | -14.310255 | 25.607173 | -10.118751 | 24.523091 | 59.359794 | -9.053347 | 11.351179 | -12.616978 |
The obtained scores for the most active and inactive pathways can be visualized as follows
dc.pl.barplot(data=pw_acts, name="treatment.vs.control", top=25, figsize=(3, 3))
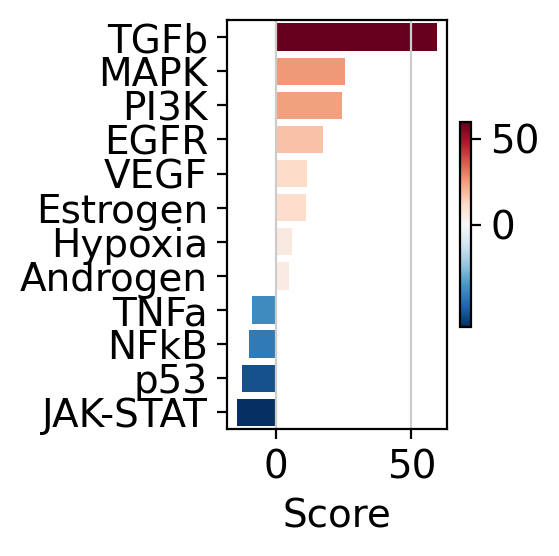
This can also be visualized using a dot plot.
# Tranform to df
df = pw_acts.melt(value_name="score").merge(
pw_padj.melt(value_name="padj")
.assign(padj=lambda x: x["padj"].clip(2.22e-16, 1))
.assign(padj=lambda x: np.log10(x["padj"]))
)
dc.pl.dotplot(df=df, x="score", y="variable", s="padj", c="score", scale=0.15, figsize=(4, 4))
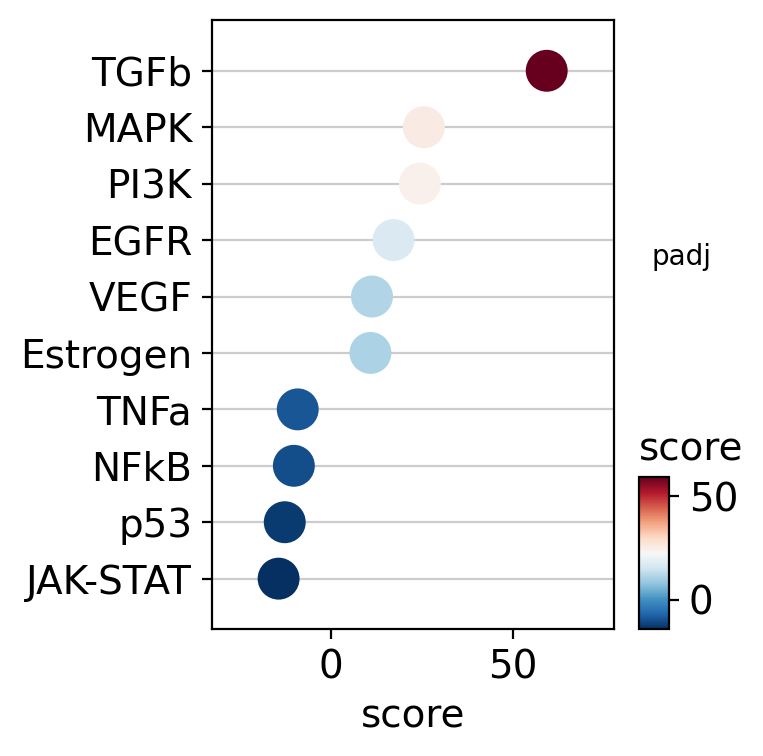
As expected, treatment with the cytokine TGF-β results in increased activity of the TGF-β pathway.
Conversely, the treatment appears to reduce the activity of other pathways, such as JAK-STAT and p53.
The targets of the TGF-β pathway can be visualized in a scatter plot.
dc.pl.source_targets(data=results_df, x="weight", y="stat", net=progeny, name="TGFb", top=15, figsize=(4, 4))
2 [-0.42468505 -0.06114804]
12 [-0.23968655 0.46555233]
2 [-0.33391819 -0.95822728]
12 [-0.88486006 -0.40560564]
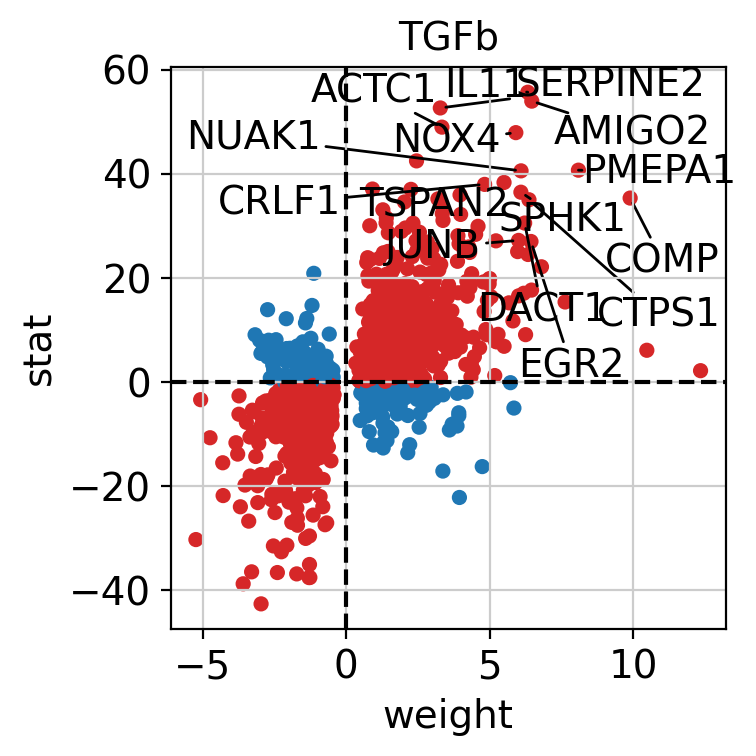
The observed activation of the TGF-β pathway is driven by the fact that most of its positively weighted target genes have positive t-values (first quadrant), while most of the negatively weighted targets have negative t-values (third quadrant).
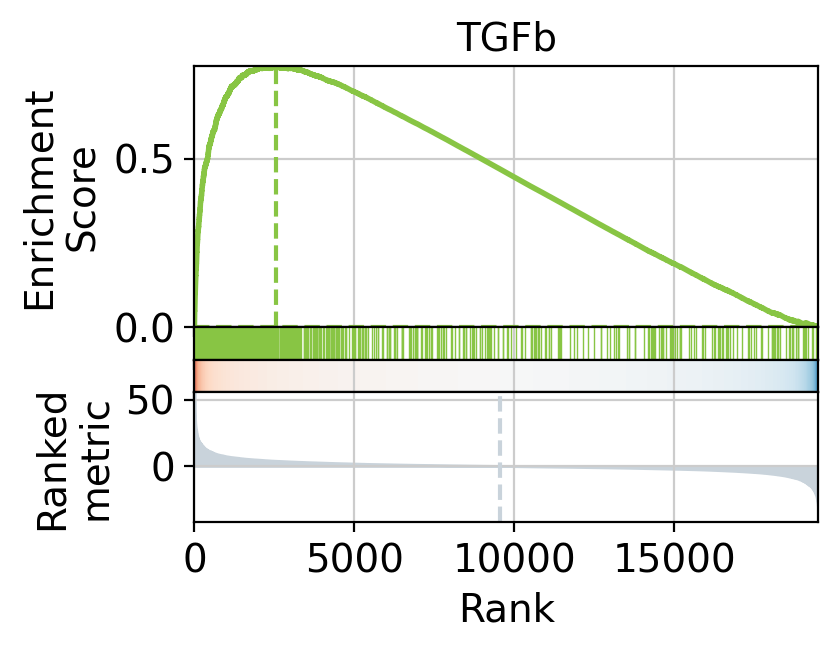
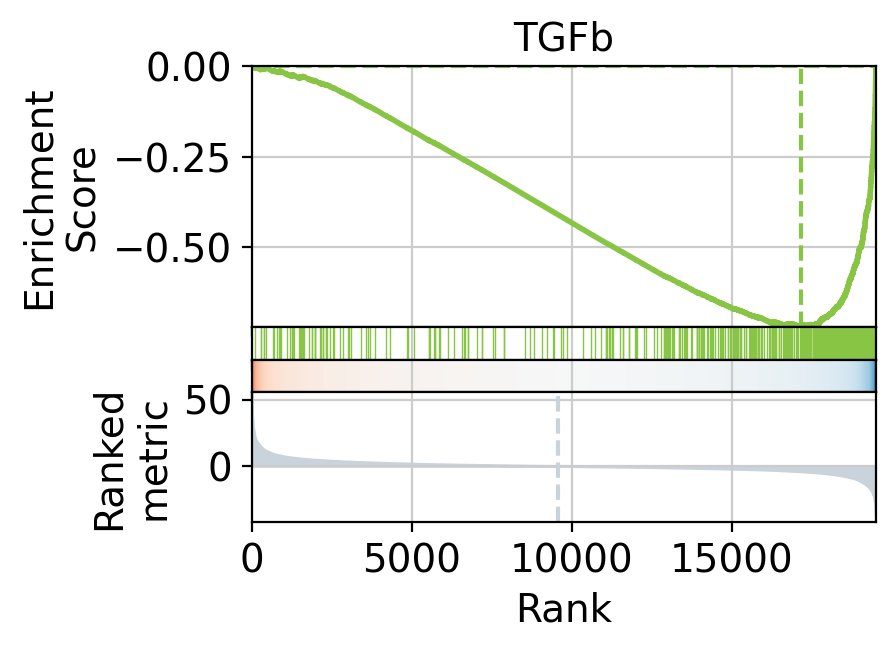
We can also visualize the targets of TGF-β as a leading edge plot. Because PROGENy includes both positive and negative target genes, the leading edge plot can be separated into positive and negative components.
_, pos_le = dc.pl.leading_edge(
results_df,
stat="stat",
net=progeny[progeny["weight"] > 0],
name="TGFb",
)
print("(+) leading edge:", pos_le[:5])
_, neg_le = dc.pl.leading_edge(
results_df,
stat="stat",
net=progeny[progeny["weight"] < 0],
name="TGFb",
)
print("(-) leading edge:", neg_le[:5])
(+) leading edge: ['IL11' 'AMIGO2' 'SERPINE2' 'ACTC1' 'NOX4']
(-) leading edge: ['SMAD3' 'ADAMTS1' 'GALNT15' 'HGF' 'NID2']
Hallmark gene sets#
Hallmark gene sets are curated collections of genes that represent specific, well-defined biological states or processes. They are part of MSigDB and were developed to reduce redundancy and improve interpretability compared to older, more overlapping gene set collections [LSP+11].
A total of 50 gene sets are provided, designed to be non-redundant, concise, and biologically coherent.
This is how to access them.
hallmark = dc.op.hallmark(organism="human")
hallmark
| source | target | |
|---|---|---|
| 0 | IL2_STAT5_SIGNALING | MAFF |
| 1 | COAGULATION | MAFF |
| 2 | HYPOXIA | MAFF |
| 3 | TNFA_SIGNALING_VIA_NFKB | MAFF |
| 4 | COMPLEMENT | MAFF |
| ... | ... | ... |
| 7313 | PANCREAS_BETA_CELLS | STXBP1 |
| 7314 | PANCREAS_BETA_CELLS | ELP4 |
| 7315 | PANCREAS_BETA_CELLS | GCG |
| 7316 | PANCREAS_BETA_CELLS | PCSK2 |
| 7317 | PANCREAS_BETA_CELLS | PAX6 |
7318 rows × 2 columns
Scoring#
Pathway scores can be easily computed by running the ulm method.
# Run
hm_acts, hm_padj = dc.mt.ulm(data=data, net=hallmark)
# Filter by sign padj
msk = (hm_padj.T < 0.05).iloc[:, 0]
hm_acts = hm_acts.loc[:, msk]
hm_acts
| ADIPOGENESIS | ALLOGRAFT_REJECTION | APICAL_SURFACE | APOPTOSIS | BILE_ACID_METABOLISM | COMPLEMENT | E2F_TARGETS | EPITHELIAL_MESENCHYMAL_TRANSITION | FATTY_ACID_METABOLISM | G2M_CHECKPOINT | ... | MYC_TARGETS_V1 | MYC_TARGETS_V2 | MYOGENESIS | P53_PATHWAY | PROTEIN_SECRETION | REACTIVE_OXYGEN_SPECIES_PATHWAY | TGF_BETA_SIGNALING | TNFA_SIGNALING_VIA_NFKB | UNFOLDED_PROTEIN_RESPONSE | XENOBIOTIC_METABOLISM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| treatment.vs.control | -2.247627 | -3.420155 | -2.708886 | -3.983905 | -2.345957 | -4.225566 | 8.116301 | 13.67402 | -2.433111 | 8.307739 | ... | 5.278134 | 2.516805 | 6.555677 | -3.532815 | 2.502763 | -6.567071 | 5.380782 | -3.504149 | 4.94476 | -4.417521 |
1 rows × 30 columns
The obtained scores for the most active and inactive pathways can be visualized as follows.
dc.pl.barplot(data=hm_acts, top=10, name="treatment.vs.control", figsize=(6, 3))

Or alternatively like this.
# Tranform to df
df = hm_acts.melt(value_name="score").merge(
hm_padj.melt(value_name="pvalue")
.assign(padj=lambda x: x["pvalue"].clip(2.22e-16, 1))
.assign(padj=lambda x: np.log10(x["pvalue"]))
)
dc.pl.dotplot(df=df, x="score", y="variable", s="padj", c="score", top=10, scale=0.05, figsize=(6.5, 4))
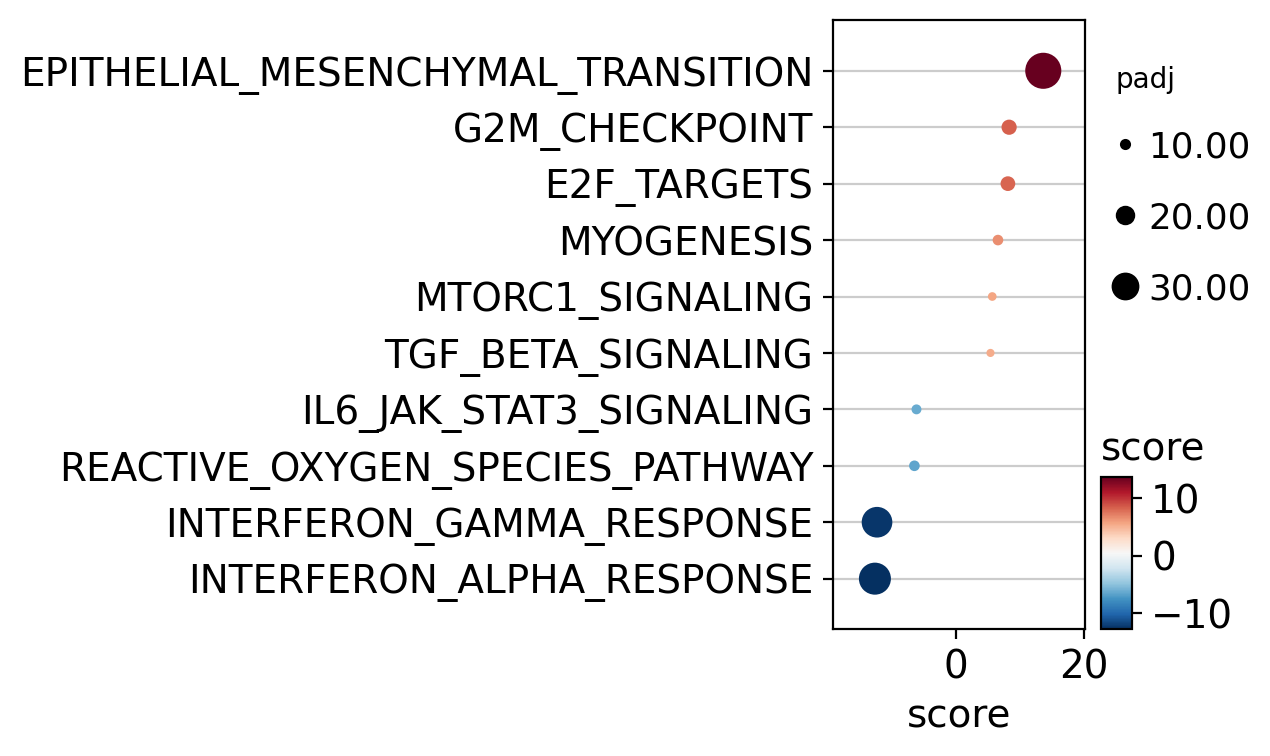
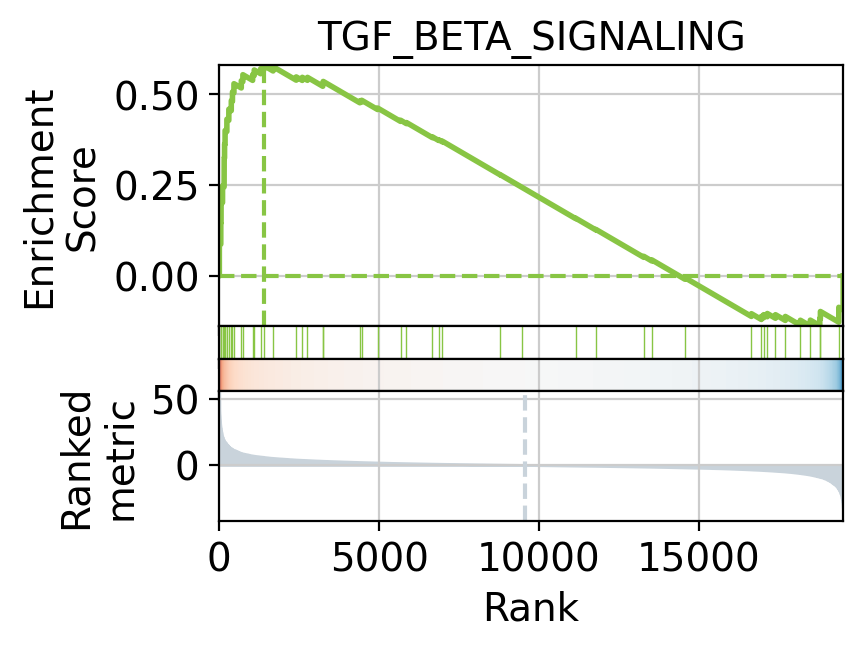
Hallmark gene sets identify TGF_BETA_SIGNALING as a positively activated significant pathway but
not as significant as EPITHELIAL_MESENCHYMAL_TRANSITION.
Targets of the TGF_BETA_SIGNALING can be visualized as a leading
edge plot.
_, le = dc.pl.leading_edge(
results_df,
stat="stat",
net=hallmark,
name="TGF_BETA_SIGNALING",
)
print("leading edge:", le[:5])
leading edge: ['PMEPA1' 'JUNB' 'SKIL' 'LTBP2' 'KLF10']